6 Two-way ANOVA
6.1 Randomised blocks of mouse treatments
Data used below are from Festing, ILAR Journal (2014) 55, 472–476 doi: 10.1093/ilar/ilu045 [1].
Four strains of mice were either used as control (C) or treated (T). We want to know whether the treatment changes the response, and whether this happens in some or all strains of mice.
The paper above gives the ANOVA table if you want to check results below.
Note that this is a fairly standard mixed-model analysis and is NOT possible in Prism 8 or 9. The description of Prism’s mixed-models is confusing and incorrect even though they claim mixed-models is same as ‘repeated measures’ and ‘within-subjects’ and ‘randomised-blocks’. See sections on Linear Mixed Effects and Block Design in Chapter 3.
Before proceeding with analysis of your own data, ensure that your data table is in the correct format and categorical variables are encoded as factors and not numbers. Here are the packages needed for these analyses.
ggplot(Mice,
aes(x = Strain,
y = GST,
group = interaction(Treatment,
Strain)))+ #note special relation declared between Treatment & Strain
geom_boxplot(aes(fill=Treatment),
width = .7,
alpha = 0.5)+
geom_point(aes(fill = Treatment,
shape = Block),
size = 3,
alpha = 0.7,
position = position_dodge(width = 0.7))+
theme_grafify()+
scale_shape_manual(values = 21:22)+ #specific shapes
labs(title = "Treatments of four strains of mice",
x = "Strain",
y = "GST activity")+
scale_fill_grafify()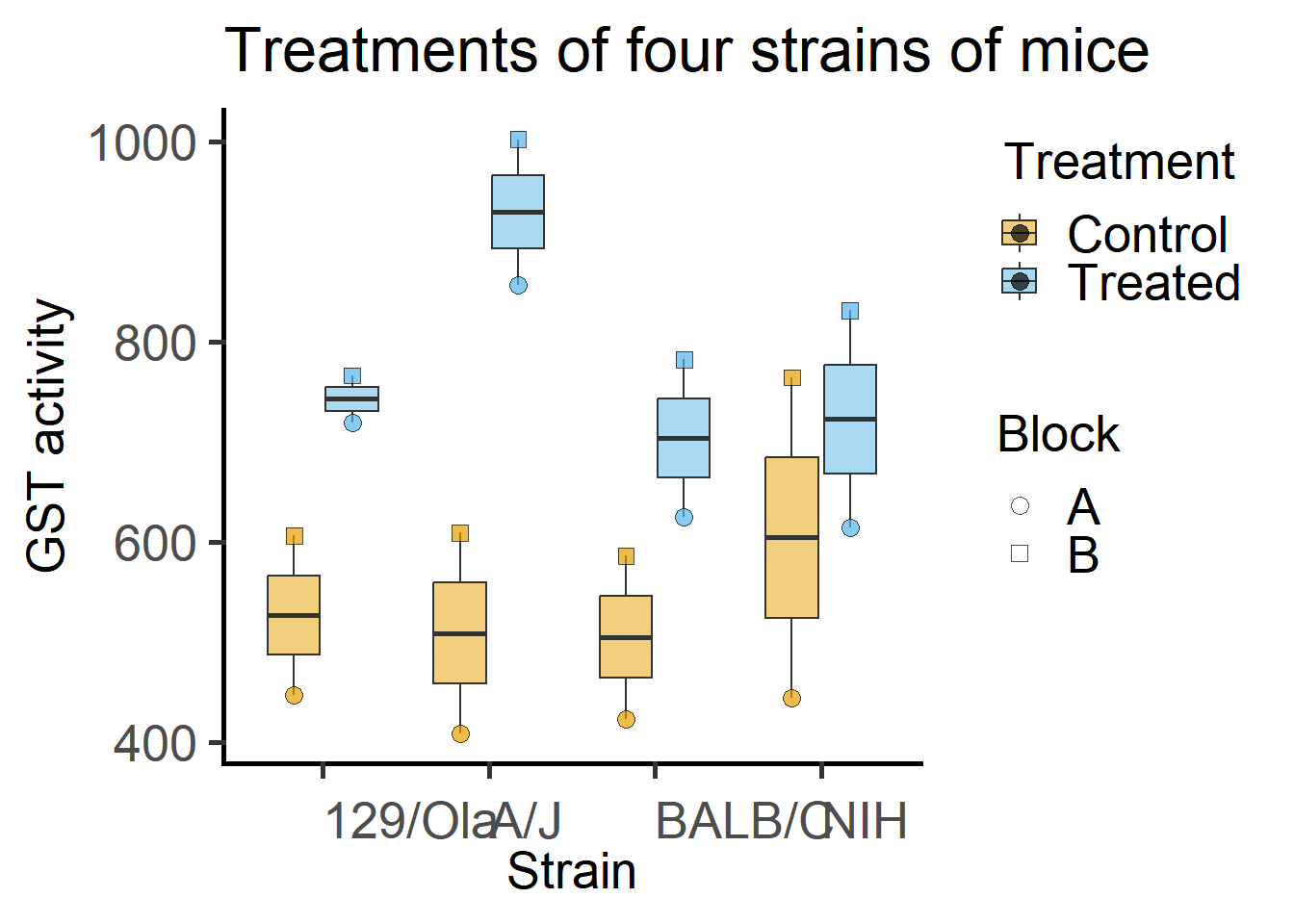
By eye-balling these data it appears that Treatment increases GST levels, but that this effect may be lesser in magnitude in the NIH strain.
We can also plot the data for each Block separately.
ggplot(Mice,
aes(x = Strain,
y = GST))+
geom_point(aes(fill = Treatment,
shape = Treatment),
alpha = 0.6,
size = 3,
position = position_dodge(width = 1))+
geom_line(aes(group = Strain),
position = position_dodge2(width = 1))+
facet_grid("Block")+
theme_grafify(base_size = 18)+
scale_fill_grafify()+
scale_shape_manual(values = 21:22)+
labs(title = "Plot of each experimental Block",
x = "Strain",
y = "GST activity")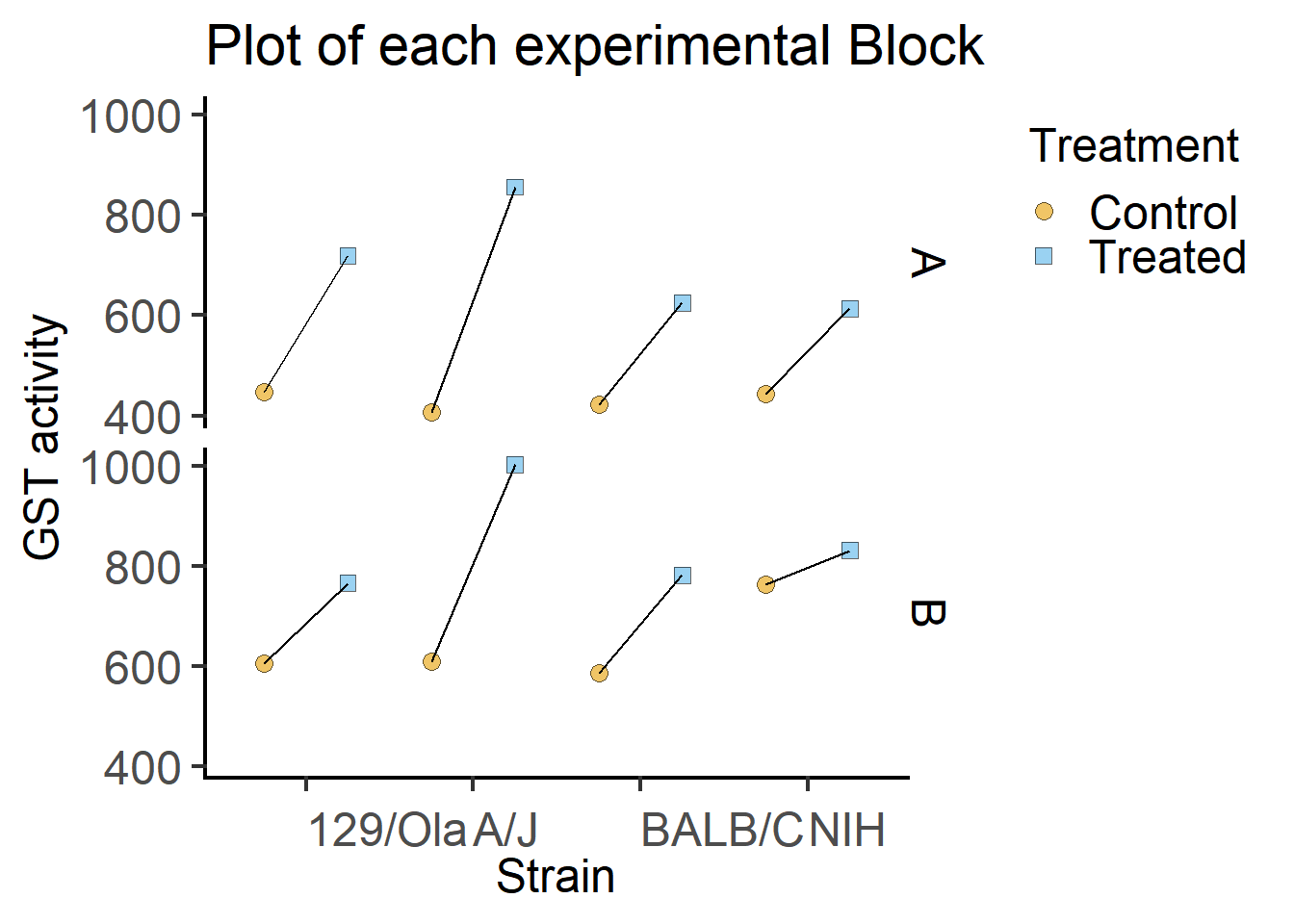
6.2 The mixed model for mouse treatments
In this data set, Y = GST activity (the dependent variable) predicted by Strain and Treatment (which are fixed factors), and Exp is the random factor.
Tip
The syntax for lme() is of the Y ~ X + (1|R) style, where Y is the dependent variable, X (one or more) are the fixed factors (the predictors) and (1|R) is the syntax for intercepts of the random factor. The quantitative variable is always on the left of the ~ and predictors on the right.
Micelmer <- lmer(GST ~ Strain*Treatment + # formula for the model
(1|Block), #random factor
data = Mice) # data tableWe can see the details of the model using the summary() call. The model and various details pop-up. The random factor details are provided separately on top. This is similar to the way we read results in Chapter 4.
An important thing to note is that in the section on Random effects which shows that the Std.Dev. of the random factor is greater than the Residual Std.Dev. Note that Std.Dev. of Block is 123.14 and the Residual is 54.38, which suggests that a large portion of \(noise\) was attributed to the random factor. An ordinary ANOVA analyses will likely be less powerful, see below.
The next part listing the number of observations (16) in two groups also looks right.
The first row in the section below gives us the overall Intercept, which by default is the alphabetically first group. (Remember you can use fct_relevel() to change order of groups). Changing the order of groups will not affect the ANOVA result!
The rest of the result shows rows with slopes (coefficients) for all combinations of levels as compared to the overall intercept. As these are linear models, they follow \(y = mx + c + \epsilon\), so each value can be calculated as slope * x + intercept \(\pm\) residual error.
summary(Micelmer) #lmer() model summaryLinear mixed model fit by REML. t-tests use Satterthwaite's method ['lmerModLmerTest']
Formula: GST ~ Strain * Treatment + (1 | Block)
Data: Mice
REML criterion at convergence: 95.9
Scaled residuals:
Min 1Q Median 3Q Max
-1.3603 -0.2462 0.0000 0.2462 1.3603
Random effects:
Groups Name Variance Std.Dev.
Block (Intercept) 15162 123.14
Residual 2957 54.38
Number of obs: 16, groups: Block, 2
Fixed effects:
Estimate Std. Error df t value Pr(>|t|)
(Intercept) 526.500 95.183 1.356 5.531 0.06830 .
StrainA/J -18.000 54.381 7.000 -0.331 0.75033
StrainBALB/C -22.000 54.381 7.000 -0.405 0.69788
StrainNIH 77.500 54.381 7.000 1.425 0.19714
TreatmentTreated 216.000 54.381 7.000 3.972 0.00538 **
StrainA/J:TreatmentTreated 204.500 76.906 7.000 2.659 0.03251 *
StrainBALB/C:TreatmentTreated -17.000 76.906 7.000 -0.221 0.83136
StrainNIH:TreatmentTreated -97.500 76.906 7.000 -1.268 0.24541
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Correlation of Fixed Effects:
(Intr) StrA/J StBALB/C StrNIH TrtmnT SA/J:T SBALB/C:
StrainA/J -0.286
StranBALB/C -0.286 0.500
StrainNIH -0.286 0.500 0.500
TretmntTrtd -0.286 0.500 0.500 0.500
StrnA/J:TrT 0.202 -0.707 -0.354 -0.354 -0.707
StBALB/C:TT 0.202 -0.354 -0.707 -0.354 -0.707 0.500
StrnNIH:TrT 0.202 -0.354 -0.354 -0.707 -0.707 0.500 0.500 This model gives us the predicted value of GST based on a mixed linear effects model fit to observed data. The observed GST values will be slightly different, but close enough if our model is good!
6.2.1 Model diagnostics
We can find out how good using model diagnostics with the ggResidpanel package. As for any linear line, the residuals should be randomly and normally distributed without any patterns. The ggResidpanel vignette is here.
resid_panel(Micelmer) #model diagnostics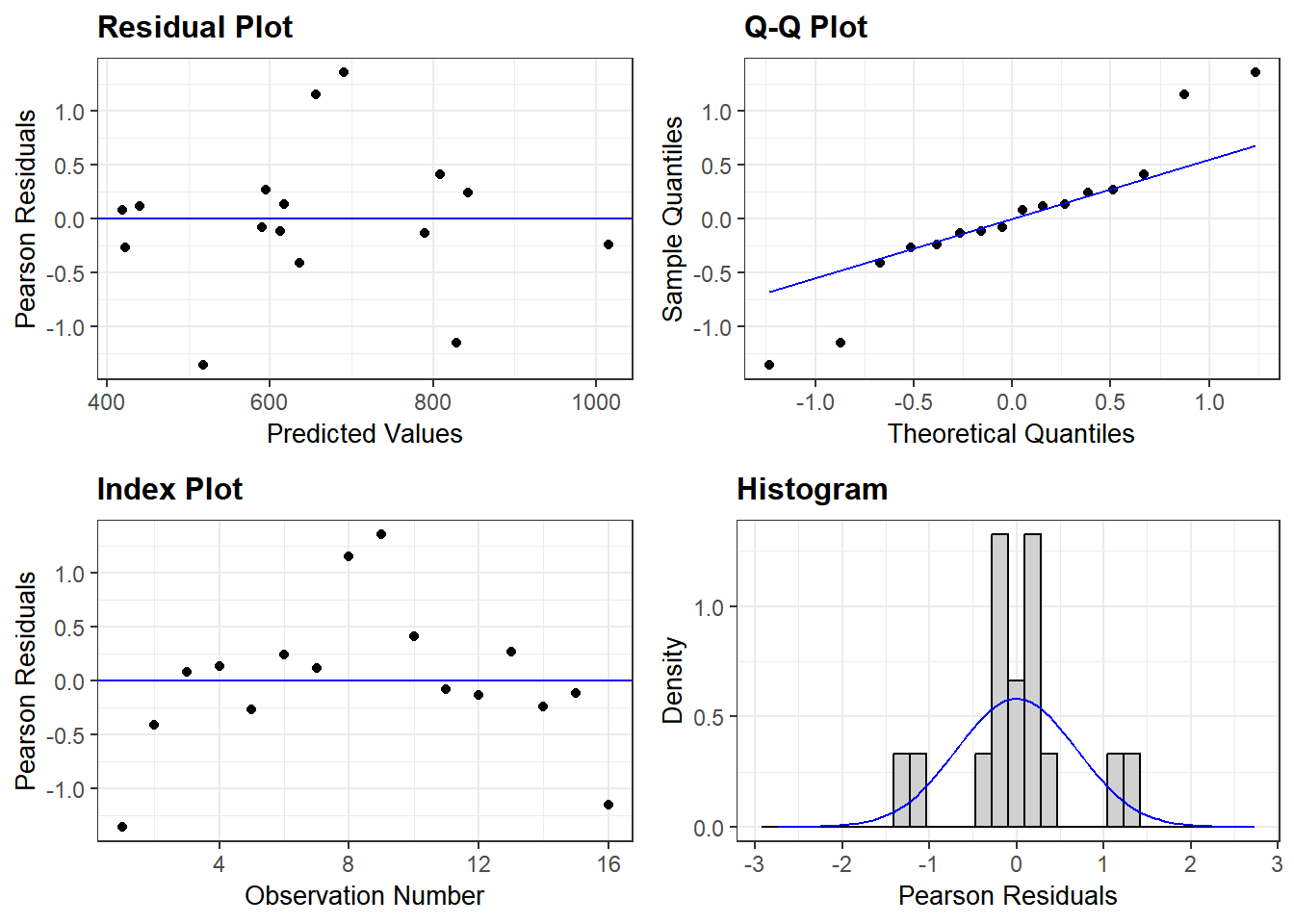
It looks like there are no major concerns with the residuals of this model. SO we can proceed with getting an ANOVA table and post-hoc comparisons.
6.2.2 ANOVA table
We will use the anova() call for the ANOVA table with the F statistic and P values. We will get P values only if lmerTest() is loaded before fitting the model. The default for lme4 package is to only provide F values (because calculating denDf for complex designs is not easy, and P values depend on numDf and denDf but F values only depend on the raw data). Like we discussed in Chapter 4, the F statistic is the ratio of the mean sum of square for each row and the residual variance of the overall model (from the summary() above).
anova(Micelmer, #anova (lmerTest)
type = "II") #get type II sum of squares - recommendedType II Analysis of Variance Table with Satterthwaite's method
Sum Sq Mean Sq NumDF DenDF F value Pr(>F)
Strain 28613 9538 3 7 3.2252 0.09144 .
Treatment 227529 227529 1 7 76.9394 5.041e-05 ***
Strain:Treatment 49590 16530 3 7 5.5897 0.02832 *
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1The F value is very large for Treatment, and relatively small for the interaction between Treatment and Strain. Strain does not seem to have a large F, but given the small \(signal\) for interaction, we might want to look at what is going on. This suggests that Treatment affects GST outcome, but the Strain of mice does not have an effect on this. The interaction suggests that the effect of Treatment on different Strains may be different.
See Chapter 5 for how to report ANOVA results. Here we would report it for each fixed factor and interaction separately.
6.3 Post-hoc comparisons of treatments
We use emmeans for this as below, which we also used here in Chapter 5. Three different post-hoc comparisons shown below as examples, but post-hoc tests should be ‘planned’ before analysis. The emmeans programme is quite powerful and allows many different options for post-hoc comparisons.
See here and here for advanced ways of setting up comparisons, and changing methods for dDf calculations etc.
6.3.1 Comparisons of Control vs Treatment for each Strain
Use the | operator with factors on each side: it could be read as pairwise comparisons of Treatment levels separately at each level of Strain.
emmeans(Micelmer,
pairwise ~ Treatment|Strain, #pairwise comparisons of Treatment at each level of strain
adjust = "fdr", #adjustment for multiple comparisons
type = "response") #back-transform (of transformation indicated in lmer() formula)$emmeans
Strain = 129/Ola:
Treatment emmean SE df lower.CL upper.CL
Control 526 95.2 1.36 -140.0 1193
Treated 742 95.2 1.36 76.0 1409
Strain = A/J:
Treatment emmean SE df lower.CL upper.CL
Control 508 95.2 1.36 -158.0 1175
Treated 929 95.2 1.36 262.5 1595
Strain = BALB/C:
Treatment emmean SE df lower.CL upper.CL
Control 504 95.2 1.36 -162.0 1171
Treated 704 95.2 1.36 37.0 1370
Strain = NIH:
Treatment emmean SE df lower.CL upper.CL
Control 604 95.2 1.36 -62.5 1270
Treated 722 95.2 1.36 56.0 1389
Degrees-of-freedom method: kenward-roger
Confidence level used: 0.95
$contrasts
Strain = 129/Ola:
contrast estimate SE df t.ratio p.value
Control - Treated -216 54.4 7 -3.972 0.0054
Strain = A/J:
contrast estimate SE df t.ratio p.value
Control - Treated -420 54.4 7 -7.733 0.0001
Strain = BALB/C:
contrast estimate SE df t.ratio p.value
Control - Treated -199 54.4 7 -3.659 0.0081
Strain = NIH:
contrast estimate SE df t.ratio p.value
Control - Treated -118 54.4 7 -2.179 0.0657
Degrees-of-freedom method: kenward-roger All versus all comparisons are also possible (not advised when there are many levels in each factor) with pairwise ~ Treatment:Strain. Note the use of : as opposed to |.
6.3.2 Comparison of the response of each Strain to the Treatment
Swapping Strain and Treatment gets us pairwise comparison of each level of Strain separately for each level of Treatment.
emmeans(Micelmer,
pairwise ~ Strain|Treatment, #pairwise comparisons of Strain
adjust = "fdr",
type = "response")$emmeans
Treatment = Control:
Strain emmean SE df lower.CL upper.CL
129/Ola 526 95.2 1.36 -140.0 1193
A/J 508 95.2 1.36 -158.0 1175
BALB/C 504 95.2 1.36 -162.0 1171
NIH 604 95.2 1.36 -62.5 1270
Treatment = Treated:
Strain emmean SE df lower.CL upper.CL
129/Ola 742 95.2 1.36 76.0 1409
A/J 929 95.2 1.36 262.5 1595
BALB/C 704 95.2 1.36 37.0 1370
NIH 722 95.2 1.36 56.0 1389
Degrees-of-freedom method: kenward-roger
Confidence level used: 0.95
$contrasts
Treatment = Control:
contrast estimate SE df t.ratio p.value
(129/Ola) - (A/J) 18.0 54.4 7 0.331 0.9004
(129/Ola) - (BALB/C) 22.0 54.4 7 0.405 0.9004
(129/Ola) - NIH -77.5 54.4 7 -1.425 0.3943
(A/J) - (BALB/C) 4.0 54.4 7 0.074 0.9434
(A/J) - NIH -95.5 54.4 7 -1.756 0.3675
(BALB/C) - NIH -99.5 54.4 7 -1.830 0.3675
Treatment = Treated:
contrast estimate SE df t.ratio p.value
(129/Ola) - (A/J) -186.5 54.4 7 -3.430 0.0220
(129/Ola) - (BALB/C) 39.0 54.4 7 0.717 0.7371
(129/Ola) - NIH 20.0 54.4 7 0.368 0.7371
(A/J) - (BALB/C) 225.5 54.4 7 4.147 0.0202
(A/J) - NIH 206.5 54.4 7 3.797 0.0202
(BALB/C) - NIH -19.0 54.4 7 -0.349 0.7371
Degrees-of-freedom method: kenward-roger
P value adjustment: fdr method for 6 tests 6.3.3 Comparison of the Treatment responses of all strains to A/J strain as an example
To get comparisons to a “control” or “reference” group, use the trt.vs.ctrl specification and set a reference group.
emmeans(Micelmer,
trt.vs.ctrl ~ Strain|Treatment, #comparison to a reference control
ref = 2, #number of reference level (alphabetical by default)
adjust = "fdr",
type = "response")$emmeans
Treatment = Control:
Strain emmean SE df lower.CL upper.CL
129/Ola 526 95.2 1.36 -140.0 1193
A/J 508 95.2 1.36 -158.0 1175
BALB/C 504 95.2 1.36 -162.0 1171
NIH 604 95.2 1.36 -62.5 1270
Treatment = Treated:
Strain emmean SE df lower.CL upper.CL
129/Ola 742 95.2 1.36 76.0 1409
A/J 929 95.2 1.36 262.5 1595
BALB/C 704 95.2 1.36 37.0 1370
NIH 722 95.2 1.36 56.0 1389
Degrees-of-freedom method: kenward-roger
Confidence level used: 0.95
$contrasts
Treatment = Control:
contrast estimate SE df t.ratio p.value
(129/Ola) - (A/J) 18.0 54.4 7 0.331 0.9434
(BALB/C) - (A/J) -4.0 54.4 7 -0.074 0.9434
NIH - (A/J) 95.5 54.4 7 1.756 0.3675
Treatment = Treated:
contrast estimate SE df t.ratio p.value
(129/Ola) - (A/J) -186.5 54.4 7 -3.430 0.0110
(BALB/C) - (A/J) -225.5 54.4 7 -4.147 0.0101
NIH - (A/J) -206.5 54.4 7 -3.797 0.0101
Degrees-of-freedom method: kenward-roger
P value adjustment: fdr method for 3 tests 6.3.4 Approx Df calculations of mouse treatments
\(signal\) Df for Strains = 4 – 1 = 3
\(signal\) Df for Treatment = 2 – 1 = 1
Interaction Signal = 3 x 1 = 3
Blocks = 2 – 1 = 1
Total samples Df = 16 – 1 = 15
\(noise\) Df = 15 – 3 – 1 – 3 – 1 = 7
Check out tutorial on Df by Ron Dotsch here.
6.4 Ordinary two-way ANOVA
Just as we did here in Chapter 5, I illustrate analyses of these data as an ordinary two-way ANOVA using lm from base R by ignoring the blocking factor. The results are different!
Micelm <- lm(GST ~ Strain*Treatment, # formula for the model
data = Mice) # data table
summary(Micelm) #model summary
Call:
lm(formula = GST ~ Strain * Treatment, data = Mice)
Residuals:
Min 1Q Median 3Q Max
-160 -80 0 80 160
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept) 526.50 95.18 5.531 0.000553 ***
StrainA/J -18.00 134.61 -0.134 0.896926
StrainBALB/C -22.00 134.61 -0.163 0.874228
StrainNIH 77.50 134.61 0.576 0.580620
TreatmentTreated 216.00 134.61 1.605 0.147239
StrainA/J:TreatmentTreated 204.50 190.37 1.074 0.314042
StrainBALB/C:TreatmentTreated -17.00 190.37 -0.089 0.931037
StrainNIH:TreatmentTreated -97.50 190.37 -0.512 0.622368
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error: 134.6 on 8 degrees of freedom
Multiple R-squared: 0.6784, Adjusted R-squared: 0.3969
F-statistic: 2.41 on 7 and 8 DF, p-value: 0.1205As we expected, the residual error is large (134.6) compared to the mixed effects model above (where it was 54.38).
I use the Anova call from the car package [2] (as the anova call only gives type I ANOVAs, it cannot be used here in a two-way ANOVA with interaction).
car::Anova(Micelm, #Anova call from "car" package
type = "II", #this is also the default
test.statistic = "F") #to get F valuesAnova Table (Type II tests)
Response: GST
Sum Sq Df F value Pr(>F)
Strain 28613 3 0.5264 0.67640
Treatment 227529 1 12.5570 0.00758 **
Strain:Treatment 49591 3 0.9123 0.47705
Residuals 144957 8
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1In this case, the result is quite different from that obtained with mixed effects model above!
emmeans(Micelm,
pairwise ~ Treatment|Strain, #pairwise comparisons of Treatments
adjust = "fdr",
type = "response")$emmeans
Strain = 129/Ola:
Treatment emmean SE df lower.CL upper.CL
Control 526 95.2 8 307 746
Treated 742 95.2 8 523 962
Strain = A/J:
Treatment emmean SE df lower.CL upper.CL
Control 508 95.2 8 289 728
Treated 929 95.2 8 710 1148
Strain = BALB/C:
Treatment emmean SE df lower.CL upper.CL
Control 504 95.2 8 285 724
Treated 704 95.2 8 484 923
Strain = NIH:
Treatment emmean SE df lower.CL upper.CL
Control 604 95.2 8 385 823
Treated 722 95.2 8 503 942
Confidence level used: 0.95
$contrasts
Strain = 129/Ola:
contrast estimate SE df t.ratio p.value
Control - Treated -216 135 8 -1.605 0.1472
Strain = A/J:
contrast estimate SE df t.ratio p.value
Control - Treated -420 135 8 -3.124 0.0141
Strain = BALB/C:
contrast estimate SE df t.ratio p.value
Control - Treated -199 135 8 -1.478 0.1776
Strain = NIH:
contrast estimate SE df t.ratio p.value
Control - Treated -118 135 8 -0.880 0.4044Note that in these analyses only one strain (A/J) shows a significant effects of Treatment on GST.
The increased power in being able to detect small differences in these variables was possible because the experiment was designed as randomised-blocks and analysed using mixed linear effects.
6.5 Two-way ANOVA with grafify
The same functions in grafify can do one or multi-way ANOVAs as linear models (simple_model or mixed_model). Please see further details on the grafify vignette website for ordinary ANOVA and mixed models.
#fit a model
graf_2w_model <- mixed_model(data = Mice,
Y_value = "GST",
Fixed_Factor = c("Strain",
"Treatment"),
Random_Factor = "Block")
#QQ plot of model residuals
plot_qqmodel(Model = graf_2w_model)
grafify#ANOVA table
mixed_anova(data = Mice,
Y_value = "GST",
Fixed_Factor = c("Strain",
"Treatment"),
Random_Factor = "Block")Type II Analysis of Variance Table with Kenward-Roger's method
Sum Sq Mean Sq NumDF DenDF F value Pr(>F)
Strain 28613 9538 3 7 3.2252 0.09144 .
Treatment 227529 227529 1 7 76.9394 5.041e-05 ***
Strain:Treatment 49591 16530 3 7 5.5897 0.02832 *
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1#posthoc comparisons at each level
posthoc_Levelwise(Model = graf_2w_model,
Fixed_Factor = c("Treatment",
"Strain"))$emmeans
Strain = 129/Ola:
Treatment emmean SE df lower.CL upper.CL
Control 526 95.2 1.36 -140.0 1193
Treated 742 95.2 1.36 76.0 1409
Strain = A/J:
Treatment emmean SE df lower.CL upper.CL
Control 508 95.2 1.36 -158.0 1175
Treated 929 95.2 1.36 262.5 1595
Strain = BALB/C:
Treatment emmean SE df lower.CL upper.CL
Control 504 95.2 1.36 -162.0 1171
Treated 704 95.2 1.36 37.0 1370
Strain = NIH:
Treatment emmean SE df lower.CL upper.CL
Control 604 95.2 1.36 -62.5 1270
Treated 722 95.2 1.36 56.0 1389
Degrees-of-freedom method: kenward-roger
Confidence level used: 0.95
$contrasts
Strain = 129/Ola:
contrast estimate SE df t.ratio p.value
Control - Treated -216 54.4 7 -3.972 0.0054
Strain = A/J:
contrast estimate SE df t.ratio p.value
Control - Treated -420 54.4 7 -7.733 0.0001
Strain = BALB/C:
contrast estimate SE df t.ratio p.value
Control - Treated -199 54.4 7 -3.659 0.0081
Strain = NIH:
contrast estimate SE df t.ratio p.value
Control - Treated -118 54.4 7 -2.179 0.0657
Degrees-of-freedom method: kenward-roger